An Automated Pipeline for Tracing Presence/Absence Variations in Eukaryotic & Prokaryotic Genomes
Introduction
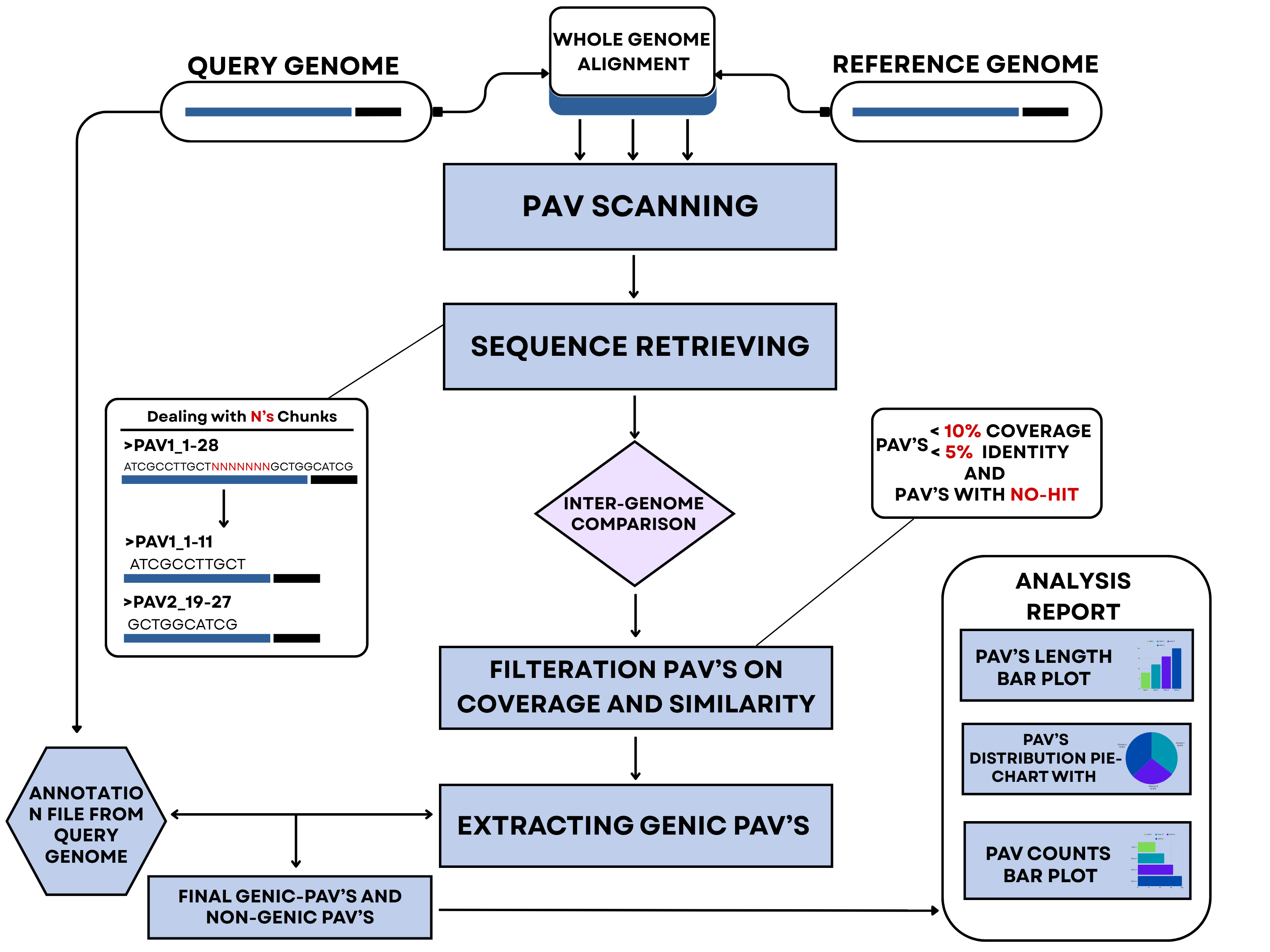
PAV (Presence/Absence Variation) is a form of structural variation where specific genes or genomic regions are present in some individuals or species but absent in others. Unlike small-scale variations like SNPs, PAVs involve larger segments of DNA and can significantly impact phenotype by altering gene content. Common in both eukaryotic and prokaryotic genomes, PAVs contribute to genetic diversity, adaptation, evolution, and traits such as disease resistance or pathogenicity. They often result from gene duplication, deletion, or horizontal gene transfer, and their analysis is crucial in fields like evolutionary biology, agriculture, microbial genomics, and personalized medicine.
sudo apt install mummer4
or build from source, you can download by clicking HERE :
wget https://github.com/mummer4/mummer/releases/download/v4.0.0beta2/mummer-4.0.0beta2.tar.gz
tar xzf mummer-4.0.0beta2.tar.gz
cd mummer-4.0.0beta2
./configure --prefix=/usr/local
make
sudo make install
export PATH=/usr/local/bin:$PATH
BLAST+:
sudo apt install ncbi-blast+
or download & extract. You can download by clicking HERE :
wget ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/ncbi-blast-*-src.tar.gz
tar xzf ncbi-blast-*-src.tar.gz
cd ncbi-blast-*/c++
# then configure & make as per NCBI docs
Bedtools
Bedtools – the swiss army knife for genome arithmetic. You can download via
Bedtools.
XtractPAV is not version specific to bedtools, however version 2.27.1 or the latest version is preferred.
sudo apt install bedtools
or
tar -zxvf bedtools-2.27.1.tar.gz
cd bedtools2
make
# Add Bedtools tools to your PATH
export PATH=/path/to/bedtools/bin:$PATH
Python Dependencies (via pip):
pip install biopython==1.78 Here
pip install plotly==6.0.1 Here
Parameters
Flag
Description
Default
--rf
Reference genome FASTA file
(required)
--ra
Reference GFF3 annotation
(required)
--qf
Query genome FASTA files (For Multi Genomes use comma‑sep)
(required)
--qa
Query GFF3 annotation files (For Multi Genomes use comma‑sep)